AWA: Academic Writing at Auckland
A Research Methods Report helps the writer learn the experimental procedures and the ways research findings are made in that discipline (Nesi & Gardner, 2012, p. 153). The question to be investigated is often provided as part of the assignment, and there is usually less focus on existing research and much more on the methods and results of the writer's own research. An IMRD (Introduction, Methods, Results, Discussion) structure is often used. AWA Research Methods Reports include Experiment Reports, Field Reports and Lab Reports.
Title: Genetic Search for Anomalous Primates Using DNA Samples
|
Copyright: Imogen Bunting
|
Description: Is this DNA sample an anomalous primate?
Warning: This paper cannot be copied and used in your own assignment; this is plagiarism. Copied sections will be identified by Turnitin and penalties will apply. Please refer to the University's Academic Integrity resource and policies on Academic Integrity and Copyright.
|
Writing features
|
Genetic Search for Anomalous Primates Using DNA Samples
|
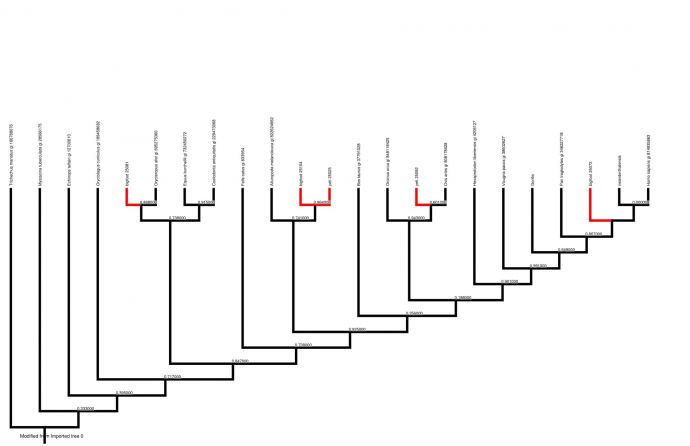
Introduction For centuries, there have been rumoured sightings of anomalous primates, or cryptids, such as the Himalayan yeti and North American sasquatch/bigfoot. These creatures are the basis of many legends, and some people have produced photographs and body parts which seem to prove their existence. There is much speculation as to whether these cryptids are real, and many theories about exactly what type of animal they are. Some have suggested that they are surviving populations of hominids thought to be extinct, or hybrids between humans and other mammals. Although eyewitness accounts exist, many scientists believe that since no bones or other solid remains of anomalous primates have been found, and no living specimens have been captured, their existence cannot be proved. As some previously undiscovered large mammals have been identified recently, it is possible that anomalous primates could exist without having been discovered by scientists (Coltman & Davies, 2006). In this experiment, our aim was to investigate whether there could possibly be a basis for the legends surrounding yeti and sasquatch. We were given five DNA samples attributed to either yeti or bigfoot. We used computer software to analyse these DNA samples and attempted to classify them within the mammalian phylogenetic tree. Previous genetic analyses of fur samples have never yet proven the existence of an anomalous primate. When Milinkovitch et al (2004) performed genetic analysis on a Nepalese fur sample, maximum parsimony, maximum likelihood and neighbour joining trees all placed it within the ungulate family, while Coltman & Davies (2006) found that when a fur sample from Canada attributed to a sasquatch was genetically analysed, it was almost genetically identical to that of a buffalo (Bison bison). Sykes et al (2014) attributed two Himalayan fur samples, supposedly from yeti, to an anomalous species of bear; however, other scientists have suggested that the fur could have come from brown bears (Guitérrez & Pine, 2015). We were interested in whether analysing our samples would produce similar results. Our null hypothesis was that none of the “yeti” or “bigfoot” DNA samples would be placed within the primate clade. Our alternative hypothesis was that at least one DNA sequence attributed to an anomalous primate would fit within the primate clade. Methods We were provided with five DNA sequence data, two attributed to yeti and three attributed to bigfoot; as well as similar nucleotide sequences from 16 other placental mammals. To facilitate comparisons, we added two more nucleotide sequences. One was a sequence for a gorilla gene which we downloaded from the NCBI website by using an accession number to find the specific sequence we wanted. The other was a Neanderthal sequence which we found using NCBI’s tool BLAST by searching for sequences that were somewhat similar to one of our bigfoot sequences. We used EBI’s MUSCLE software to align our 23 sequences to ensure that we were comparing homologous regions of DNA. Our aligned sequences were imported into the data organization and analysis program Mesquite. Here, we could view a character matrix of our aligned sequences (Figure 1, page 3). Some regions of DNA had to be cut out because they did not align properly. The gorilla nucleotide sequence from BLAST was a complete sequence as opposed to a fragment like the other sequences, so there were large regions at the ends which had to be deleted because they did not align with any other mammals. Also, at some loci so many mutations had occurred it was difficult to determine whether the characters were aligned properly, so these ambiguous regions were deleted. The edited character matrix was exported to PhyML, a computer program that estimates phylogenetic trees. We generated a phylogenetic tree using maximum likelihood (Figure 2, page 4). This tree was edited using Mesquite. The manatee (Trichechus manatus) was selected as the outgroup because it is not closely related to any of the other placental mammals used for this experiment, and choosing the manatee as the outgroup meant that all the primates were grouped together at one end, making the tree easier to interpret. Branches leading to supposed anomalous primates are highlighted in red.
Results The branches of the phylogenetic tree (Figure 2, page 4) which lead to suspected anomalous primates are located at various positions throughout the tree. Two of the anomalous primates (a yeti and a bigfoot) form their own polyphyletic clade, which appears as a sister taxon to the giant panda (Ailuropoda melanoleuca). The other yeti was placed firmly within the ungulate clade, as a sister taxon to the sheep (Ovis aries). One bigfoot was positioned as a sister taxon to the aardvark (Orycteropus afer). The other bigfoot was actually positioned within the primate clade, as a sister taxon to recent hominids (Homo neanderthalensis and the modern human, Homo sapiens). There are varying degrees of uncertainty at the nodes of the tree as some branches are more probable than others. Discussion Before making judgements about the validity of our results, we must consider their accuracy and precision. MUSCLE is probably the most accurate multiple sequence alignment tool currently available (Edgar, 2004), and ambiguous regions of our DNA matrix were removed, so we can be reasonably sure that the DNA alignment matrix is accurate. There is a greater measure of uncertainty around the phylogenetic tree. Maximum likelihood is only one method of estimating phylogeny, and although it is reasonably accurate, the accuracy of our results could possibly have been improved if we had used other methods as well and generated a consensus tree. In addition, the probabilities at the internal nodes of the tree vary greatly. For instance, the probability at the node between Homo sapiens and Homo neanderthalensis is 0.000000, so we can be almost certain that this branch is accurate, but many other branches of the tree have statistically insignificant probabilities. In addition, we only compared the sequences of a single DNA fragment. If we had been able to compare larger fragments, or multiple genes, the results would have been more reliable; however, this would have been much more time-consuming. If we assume that the tree is reasonably accurate, it presents some interesting findings. Four of the DNA samples we investigated are clearly not from anomalous primates. The fact that one of them came from an ungulate is in accordance with Milinkovitch et al (2004) and Coltman & Davies (2006). The two sequences which formed a sister taxon to giant panda could perhaps be similar to those which Sykes et al (2014) attributed to an “anomalous bear”. The bigfoot DNA sequence which appears to belong to a primate presents a less straightforward case. It seems to be a closer relative of modern humans than the common chimpanzee (Pan troglodytes), but a more distant relative than the Neanderthal. This suggests that the DNA could have come from either an earlier hominid, or another great ape such as a bonobo. We reject our null hypothesis in favour of the alternative hypothesis – one of our uncertain DNA samples does belong to a primate. It would be advisable to run a similar experiment comparing this DNA sequence with homologous sequences from extinct hominids and other apes in order to try to find a match and ascertain whether or not this really is evidence that the sasquatch exists.
References Coltman D, Davis C. 2006. Molecular cryptozoology meets the Sasquatch. Trends Ecol Evol 21: 60-61 Edgar R. 2004. Muscle: multiple sequence alignment with high accuracy and high throughput. Nucleic Acid Res 32: 1792-1797 Guitérrez E, Pine RH. 2015. No reason to believe that Sykes’s Yeti-bear cryptid exists. Skeptical Observer 39: 29 Milinkovitch MC, Caccone A, Amato G. 2004. Molecular phylogenetic analyses indicate extensive morphological convergence between the “yeti” and primates. Mol Phylogenet Evol 31: 1-3 Sykes BC, Mullis RA, Hagenmuller C, Melton TW, Sartori M. 2014. Genetic analysis of hair samples attributed to yeti, bigfoot and other anomalous primates. Proc R Soc B 281: 20140161 |
|